Fenilketonuria
Marian Grosser humán orvostudományt tanult Münchenben. Ezenkívül a sok minden iránt érdeklődő orvos izgalmas kitérőket merészelt tenni: filozófiát és művészettörténetet tanulni, rádión dolgozni, végül pedig egy Netdoctornak is.
További információ a szakértőiről A összes tartalmát orvosi újságírók ellenőrzik.A fenilketonuria (PKU) a fehérje -anyagcsere veleszületett, örökletes betegsége. Megakadályozza a fenilalanin aminosav lebomlását. Ez felhalmozódik a szervezetben, és megzavarja a gyermek agyának fejlődését. Ha nem kezelik, a fenilketonuria súlyos értelmi fogyatékossághoz vezet. Időben történő terápiával azonban a betegek normális életet élhetnek. Tudjon meg mindent a fenilketonuriáról itt!
Ennek a betegségnek az ICD -kódjai: Az ICD -kódok nemzetközileg elismert orvosi diagnosztikai kódok. Megtalálhatók például az orvos leveleiben vagy a keresőképtelenségi igazolásokon. E70
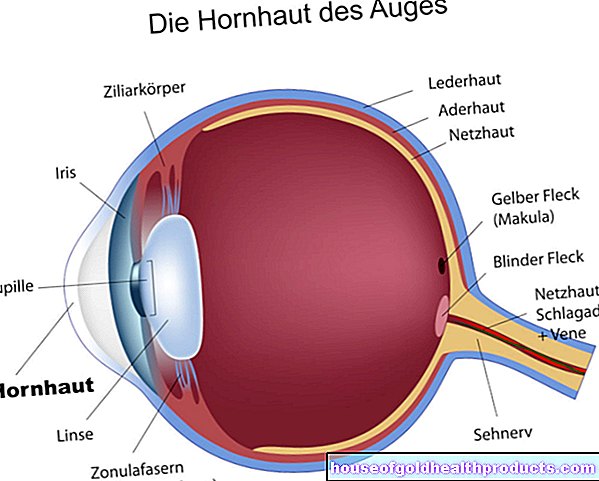
Fenilketonuria: leírás
A fenilketonuria (PKU) egy öröklött anyagcsere -betegség, amely születésétől fogva fennáll, és zavarja az esszenciális aminosav -fenilalanin lebomlását. Az aminosavak a fehérjék alapvető építőkövei, és így létfontosságú anyagcsere -összetevőik. Némelyikük csak táplálékkal juthat be a szervezetbe, a szervezet maga nem tudja előállítani őket. Az ilyen aminosavakat esszenciálisnak nevezik.
Mi történik a fenilketonuriával?
Általában az aminosavak egyensúlyban vannak az abszorpció / felhalmozódás és a lebomlás között, így mindig annyi rendelkezésre áll, amennyi a szervezetnek szüksége van. Különböző aminosavak esetében a hiány vagy a túlzott mennyiség jelentős kárt okozhat és különböző tüneteket okozhat.
Az úgynevezett klasszikus PKU-ban a fenilalanin-hidroxiláz (PAH) hatása korlátozott, vagy teljesen hiányzik. A PAH -hiány miatt a fenilalanin egyre inkább felhalmozódik a szervezetben. A túl magas fenilalanin -koncentráció jelentősen megzavarja az agy fejlődését, és korai stádiumban értelmi fogyatékossághoz vezet a fiatal betegeknél.
Mivel a betegség esetén a fenilalanin normális lebomlása nem lehetséges, más bomlástermékek, az úgynevezett fenil-ketonok képződnek. A vizelettel ürülnek, és felelősek a betegség nevéért.
Atipikus fenilketonuria
Még a fenilketonuria atipikus formái esetén is zavart okoz a fenilalanin lebomlása. Ennek oka azonban nem a PAH hibája. Ehelyett egy koenzim, a tetrahidrobiopterin (BH4) funkciója korlátozott. Közvetve részt vesz a fenilalanin lebontásában, mivel a PAH -nak szüksége van BH4 -re ahhoz, hogy a fenilalanint tirozinná alakítsa.
Mivel a BH4 a dopamin és a szerotonin hírvivő anyagok előállítása szempontjából is fontos, az atipikus, BH4 -hiányos fenilketonuria általában bonyolultabb, mint a klasszikus forma.
Kit érint a fenilketonuria?
A PKU az egyik leggyakoribb veleszületett anyagcsere -betegség. Becslések szerint világszerte körülbelül minden hetedik újszülött fogja kifejleszteni, nincs különbség lányok és fiúk között. Mivel ez örökletes betegség, a család több tagja gyakran érintett.
Phenyketonuria: tünetek
Eleinte a fenilketonuriában szenvedő csecsemők nem mutatnak semmilyen betegségtünetet. Az első problémák csak az élet negyedik -hatodik hónapjában jelentkeznek, ha a betegséget még nem ismerték fel és nem kezelték. Mindenekelőtt a zavart agyérés idővel hatalmas szövődményeket okoz. A kezeletlen PKU tünetei a következők:
- erős mentális hiány. Az agykárosodás a pubertás során előrehalad, majd stagnál. Az érintett gyermekek ilyenkor általában súlyosan értelmi fogyatékosok.
- Görcsrohamok (epilepszia). A károsodás miatt az agy idegsejtjei különösen érzékenyek és túlgerjeszthetők. Gyakori epilepsziás rohamok az eredmény.
- motoros fogyatékosság. Nemcsak az agysejtek, hanem a beteg izmai is izgatottak lehetnek. Ezért gyakran feszültté válik (görcsösség), ami különböző mozgászavarokhoz vezet.
- Viselkedési zavarok. Néhány fenilketonuriás gyermek hiperaktív és szokatlanul agresszív, és a haragkitörések is gyakoribbak.
- egy kis fej (mikrocefália). Mivel a beteg agya nem fejlődik megfelelően, a fej növekedése is elmarad. A kis fejkörfogat társaikhoz képest különösen az idősebb gyermekeknél figyelhető meg.
- észrevehető szag. A PKU bizonyos fenilalanin bomlástermékeket állít elő, amelyek illata hasonló az egér ürülékéhez. Ezek az anyagok elsősorban a vizelettel, de részben a bőrön keresztül is kiválasztódnak.
- ekcéma-szerű bőrváltozások
Mivel a fenilketonuriában a melanin pigment termelése is zavart szenved, sok beteg nagyon világos, napérzékeny bőre és fehér szőke haja van. A szem írisze is világoskék vagy átlátszó, és lehetővé teszi a vöröses szemfenék átvilágítását.
A PKU tünetei személyenként eltérőek. Ennek fő oka az, hogy a fenilalanin -hidroxiláz (PAH) aktivitása minden egyes betegnél eltérően korlátozott. Néhányuk még mindig rendelkezik bizonyos maradék aktivitással, így kevesebb fenilalanin halmozódik fel a szervezetben. Mások egyáltalán nem mutatnak enzimaktivitást - a betegség ennek megfelelően gyorsabban és komolyabban halad.
Fenilketonuria: okok és kockázati tényezők
A fenilketonuria örökletes betegség. Ma már számos genetikai mutáció ismert, amelyek a PAH hibájához vezetnek. A mutáció típusa határozza meg, hogy a fenilalanin lebontása milyen mértékben korlátozott.
A PKU recesszíven öröklődik, ami azt jelenti, hogy egy személy a megváltozott gén hordozója lehet a betegség kialakulása nélkül. Hasonlóképpen, a fenilketonuriában szenvedők egészséges gyermekeket szülhetnek.
Csak akkor van bizonyos valószínűsége, hogy utódaikban fenilketonuria alakul ki, ha mindkét szülő genetikai összetételében mutációk vannak. Ha a szülők nemcsak a gének hordozói, hanem maguk is rendelkeznek PKU -val, akkor minden gyermek együtt is kifejleszti azt.
Fenilketonuria: vizsgálatok és diagnózis
Mivel a fenilketonuria súlyos következményei megelőzhetők a kezelés időben történő megkezdésével, különösen fontos a betegség mielőbbi felfedezése. Németországban a gyermekeket a születés utáni harmadik napon általános vizsgálat (újszülött szűrés) keretében különböző veleszületett betegségek, köztük a PKU szempontjából vizsgálják.
Tandem tömegspektrometria
Sok veleszületett anyagcserezavar diagnosztizálható az úgynevezett tandem tömegspektrometria segítségével. Lehetővé teszi az újszülött vérének gyors és egyszerű vizsgálatát. A fenilketonuria mellett az orvosok több mint 20 egyéb betegséget is észlelhetnek néhány percen belül.
Guthrie teszt
A feltalálójáról elnevezett Guthrie -teszt lehetővé teszi a PKU diagnosztizálását is. Ehhez kis mennyiségű vért vesznek a gyermek sarkából, és szűrőpapírra helyezik. A laboratóriumban ezután meghatározhatja, hogy a fenilalanin koncentrációja megnőtt -e.
A Guthrie -tesztet az 1960 -as években vezették be, és régóta a standard módszer a fenilketonuria diagnosztizálására. Ennek azonban hátrányai vannak a tandem tömegspektrometriához képest. A Guthrie -teszt csak öt nap múlva ad eredményt, ami szintén hajlamos a hibákra. Például olyan tényezők, mint a gyermek étrendje vagy az esetleges antibiotikum -terápia meghamisítják az eredményeket. Néhány országban még mindig használják a Guthrie -tesztet, Németországban általában már nem.
Egyéb tesztek
Ha az újszülött szűrés során fenilketonuria gyanúja derül ki, további vizsgálat következik megerősítés céljából. Ezzel is megállapítható a vérben a fenilalanin pontos koncentrációja.
Végül fontos megkülönböztetni, hogy tipikus (klasszikus) vagy atipikus PKU. Ehhez speciális tesztek is rendelkezésre állnak, például a tetrahidrobiopterin stresszteszt. A megkülönböztetés azért fontos, mert az atipikus fenilketonuriát a klasszikus formától eltérően kezelik.
A magzatvíz vizsgálat
A fenilketonuria már a terhesség alatt is diagnosztizálható (prenatális diagnózis). Ehhez kis mennyiségű magzatvizet vesznek ki az anya magzatzsákjából, és megvizsgálják a születendő gyermek sejtjeit. A PKU -t okozó genetikai hibák azonosíthatók ilyen módon.
Újszülött szűréssel azonban a fenilketonuriát általában elég korán felfedezik a kezeléshez. Mivel a magzatvíz -vizsgálat mindig bizonyos kockázatokkal jár, a PKU diagnosztizálásához való alkalmazása általában nem hasznos.
Fenilketonuria: kezelés
A PKU -ban lévő fenilalanin -felesleg ellen csak egy módja van: Az érintett gyermekeknek speciális étrendet kell követniük annak érdekében, hogy a lehető legkevesebb fenilalanint vehessék be étkezésükkel. Szükségük van bizonyos táplálék -kiegészítőkre is, hogy helyettesítsék azokat az anyagokat, amelyek fenilalaninból képződnének egészséges gyermekeknél.
A terápiát a fejlődési rendellenesség első tüneteinek megjelenése előtt kell elkezdeni, azaz az élet első két hónapjában. A már bekövetkezett agykárosodást nem lehet visszafordítani.
Táplálkozási terápia a PKU diétával
Minden természetes fehérje a fenilalanin aminosav körülbelül öt százalékát tartalmazza. A legtöbb élelmiszer sokkal többet tartalmaz belőle, mint amennyire a szervezetnek szüksége van. Ez nem jelent problémát egy egészséges ember számára, mert lebomlik és kiválasztja a felesleges mennyiségű fenilalanint.
A fenilketonuriában szenvedő betegeknek viszont nagyrészt nélkülözniük kell a természetes fehérjéket. Az iparilag gyártott speciális termékek pótolják a hiányzó élelmiszer -összetevőket. Egyrészt meg akarjuk akadályozni a fenilalanin feleslegét, másrészt a beteget olyan anyagokkal kell ellátni, amelyek egyébként hiányoznának, például a tirozinnal, amely fenilalaninból képződik.
A PKU diéta célja azonban nem a fenilalanin felszívódásának teljes leállítása. Mivel a szervezetnek szüksége van bizonyos mennyiségű aminosavra a fontos anyagcsere -folyamatokhoz. A megfelelő koncentráció elérése nagy kihívást jelent az orvosok és a betegek számára, és sok fegyelmet igényel.
Az étrendet tehát egy életen át, de különösen szigorúan hat éves korig kell betartani. Mivel az agy nagyon erősen fejlődik egészen addig a korig, és ezért különösen érzékeny a károsodásra. Felnőtt korban a fenilalanin magas koncentrációja nem okoz agykárosodást, mint a gyermekeknél. Ezek azonban más neurológiai panaszok kiváltó okai lehetnek, például gyenge koncentráció vagy lelassult reakciók.
A fenilketonuria diétás kezelését a metabolikus betegségek speciális központjában kell elkezdeni. Mivel nem minden klinikán lehet utasítani a szülőket az étrendre. Ezt legjobban diétás tanácsadás segítségével lehet megtenni, amely bemutatja, hogyan kell követni az étrendet, és rendszeresen ellenőrizni kell a vér fenilalaninszintjét.
Az atipikus fenilketonuria kezelése
A PKU atipikus formáiban a hiányzó BH4 koenzimet és bizonyos hírvivő anyagokat, például a dopamint és a szerotonint mesterségesen pótolják. Bizonyos esetekben a betegnek alacsony fenilalanin tartalmú étrendet is követnie kell.
Fenilketonuria terhesség alatt
Ha Ön terhes, és maga is fenilketonuriában szenved, fontolja meg a következőket:
- Tartsa be különösen szigorúan étrendjét!
- Rendszeres időközönként ellenőriztesse a fenilalanin -értékeket. Mivel a születendő gyermekben a fenilalanin koncentrációja körülbelül kétszer akkora, mint a terhes nőnél. Az agy mellett károsíthatják a születendő gyermek szívét és szemét is. A gyermek csontvázának rendellenességei a terhesség alatti magas fenilalaninszint miatt is kialakulhatnak.
- Annak érdekében, hogy kizárják a gyermek agykárosodását a terhesség korai szakaszában, a fenilketonuriában szenvedő nőknek gondosan meg kell tervezniük a terhességet, és eleve óvintézkedéseket kell tenniük.
- A fenilketonuriában szenvedő terhes nőknél a szigorúan nem betartott étrend vetéléshez (spontán vetéléshez) vezethet.
- Mindenesetre a fenilketonuriában szenvedő terhes nőknek tanácsot és útmutatást kell kérniük az orvostól.
Fenilketonuria: a betegség lefolyása és prognózisa
Ha a fenilketonuriát korán diagnosztizálják, ha lehetséges, az újszülöttnél, és ha a speciális PKU -diétát követik, akkor a prognózis általában jó. A gyerekek szellemileg fejlődnek, és átlagos élettartamuk van.
Ha azonban nem kezelik, az agykárosodás súlyos mentális fejlődési rendellenességekhez vezet, amelyeket később nem lehet orvosolni. Az érintettek várható élettartama normális, de intelligencia hányadosuk szinte mindig nagyon messze van a normától.
Különös eset a fenilketonuria ritka, atipikus formája, amelyben BH4 -hiány van jelen. A fenilketonuria ezen változata progresszív idegrendszeri károsodáshoz vezethet súlyos görcsökkel az étrend ellenére.
Címkék: laboratóriumi értékek tcm bőr